Human Spermatogenic Failure Purges Deleterious Mutation Load from the Autosomes and Both Sex Chromosomes, including the Gene DMRT1
Alexandra M. Lopes, Kenneth I. Aston, Emma Thompson, Filipa Carvalho, João Gonçalves, Ni Huang, Rune Matthiesen, Michiel J. Noordam, Inés Quintela, Avinash Ramu, Catarina Seabra, Amy B. Wilfert, Juncheng Dai, Jonathan M. Downie, Susana Fernandes, Xuejiang Guo, Jiahao Sha, António Amorim, Alberto Barros, Angel Carracedo, Zhibin Hu, Matthew E. Hurles, Sergey Moskovtsev, Carole Ober, Darius A. Paduch, Joshua D. Schiffman, Peter N. Schlegel, Mário Sousa, Douglas T. Carrell, Donald F. Conrad, 21.03.2013
Abstract
Gonadal failure, along with early pregnancy loss and perinatal death, may be an important filter that limits the propagation of harmful mutations in the human population. We hypothesized that men with spermatogenic impairment, a disease with unknown genetic architecture and a common cause of male infertility, are enriched for rare deleterious mutations compared to men with normal spermatogenesis. After assaying genomewide SNPs and CNVs in 323 Caucasian men with idiopathic spermatogenic impairment and more than 1,100 controls, we estimate that each rare autosomal deletion detected in our study multiplicatively changes a mans risk of disease by 10% (OR 1.10 [1.04–1.16], p<2×10−3), rare X-linked CNVs by 29%, (OR 1.29 [1.11–1.50], p<1×10−3), and rare Y-linked duplications by 88% (OR 1.88 [1.13–3.13], p<0.03). By contrasting the properties of our case-specific CNVs with those of CNV callsets from cases of autism, schizophrenia, bipolar disorder, and intellectual disability, we propose that the CNV burden in spermatogenic impairment is distinct from the burden of large, dominant mutations described for neurodevelopmental disorders. We identified two patients with deletions of DMRT1, a gene on chromosome 9p24.3 orthologous to the putative sex determination locus of the avian ZW chromosome system. In an independent sample of Han Chinese men, we identified 3 more DMRT1 deletions in 979 cases of idiopathic azoospermia and none in 1,734 controls, and found none in an additional 4,519 controls from public databases. The combined results indicate that DMRT1 loss-of-function mutations are a risk factor and potential genetic cause of human spermatogenic failure (frequency of 0.38% in 1306 cases and 0% in 7,754 controls, p = 6.2×10−5). Our study identifies other recurrent CNVs as potential causes of idiopathic azoospermia and generates hypotheses for directing future studies on the genetic basis of male infertility and IVF outcomes.
LOPES, Alexandra M., et al. Human spermatogenic failure purges deleterious mutation load from the autosomes and both sex chromosomes, including the gene DMRT1. PLoS genetics, 2013, 9. Jg., Nr. 3, S. e1003349.
Publication: https://doi.org/10.1371/journal.pgen.1003349
 Disclaimer
Disclaimer
The publication Human Spermatogenic Failure Purges Deleterious Mutation Load from the Autosomes and Both Sex Chromosomes, including the Gene DMRT1 by Alexandra M. Lopes, Kenneth I. Aston, Emma Thompson, Filipa Carvalho, João Gonçalves, Ni Huang, Rune Matthiesen, Michiel J. Noordam, Inés Quintela, Avinash Ramu, Catarina Seabra, Amy B. Wilfert, Juncheng Dai, Jonathan M. Downie, Susana Fernandes, Xuejiang Guo, Jiahao Sha, António Amorim, Alberto Barros, Angel Carracedo, Zhibin Hu, Matthew E. Hurles, Sergey Moskovtsev, Carole Ober, Darius A. Paduch, Joshua D. Schiffman, Peter N. Schlegel, Mário Sousa, Douglas T. Carrell, Donald F. Conrad is published under an open access no derivatives license: : https://creativecommons.org/licenses/by/4.0/. Permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Curation by the MFGA team Relevant data sets presented in the publication have been identified. If possible, annotations (title, general information, conditions, processed tissue types and processed cell types) have been added based on information from the publication. Data tables and images that provide a good overview on the publication's findings on the data set have been extracted from the publication and/or supplement. If not stated otherwise, images are depicted with title and description exactly as in the publication. Tables have been adjusted to the MFGA table format. Conducted adjustments are explained in the detailed view of the tables. However, titles and descriptions have been adopted from the publication.
Data set 1: Studying the genomewide distribution of CNVs in our primary cohort from Utah
Genome: Genome-wide Association Study
Species
| Species |
|---|
| Human |
Conditions
| Human phenotype ontology | Participants | Comment |
|---|---|---|
| HP:0011961: Non-obstructive azoospermia | 35 | idiopathic non-obstructive azoospermia; men from Utah |
| HP:0000798: Oligozoospermia | 48 | men from Utah |
| HP:control | 62 | men from Utah |
Tissue Types
| BRENDA tissue ontology | Maturity | Description | Species | Replicates |
|---|---|---|---|---|
| BTO_0000089: blood | Adult | Human |
Cell Types
| Cell ontology | Maturity | Description | Species | Replicates | Cells per replicate |
|---|---|---|---|---|---|
| CL_0000542: Lymphocytes | Adult | Peripheral blood lymphocytes | Human |
Images
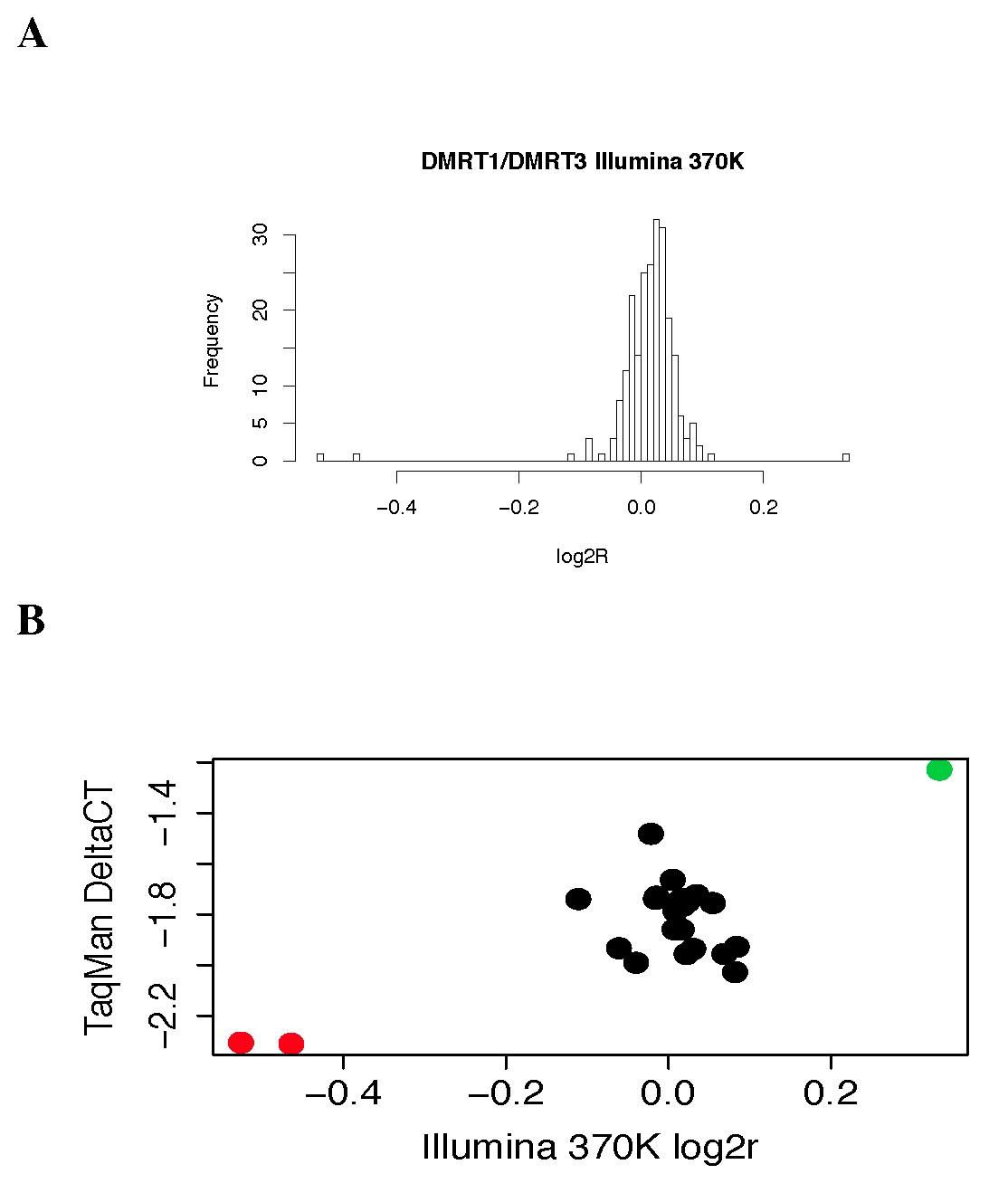
Figure S10: qPCR validation of DMRT1 deletions in the Utah Cohort
A: Histogram of mean probe intensities from Illumina 370K array spanning the DMRT1/DMRT3 deletion locus (chr9:845901–994958 bp). N = 148 samples are plotted.<br/>B: Taqman validation results for the DMRT1 locus from 30 of the samples screened by 370K array in panel A (y-axis), including the two deletion carriers (red points) and one duplication carrier (green point) identified from Illumina 370K intensity data (x-axis).
Licensed under: https://creativecommons.org/licenses/by/4.0/
Figure S2 A: Principal components analysis (PCA) of population structure in Utah cohort post-QC
Samples were analyzed together with HapMap samples using the EIGENSOFT package. Eigenvector loadings for cases and controls are plotted as red crosses, while HapMap samples are plotted as other colored symbols described in each legend.
Licensed under: https://creativecommons.org/licenses/by/4.0/
Data set 2: Replication of the initial findings in Caucasion Cohort (Cohort 2-Porto, Weill Cornell)
Genome: Microarray
Species
| Species |
|---|
| Human |
Conditions
| Human phenotype ontology | Participants | Comment |
|---|---|---|
| HP:0000027: Azoospermia | 179 | Caucasian men with idiopathic azoospermia |
| HP:control | 368 | unphenotyped control set of Spanisch men |
Tissue Types
| BRENDA tissue ontology | Maturity | Description | Species | Replicates |
|---|---|---|---|---|
| BTO_0000089: blood | Adult | human |
Cell Types
| Cell ontology | Maturity | Description | Species | Replicates | Cells per replicate |
|---|---|---|---|---|---|
| CL_0000542: Lymphocytes | Adult | Peripheral blood lymphocytes | Human |
Images
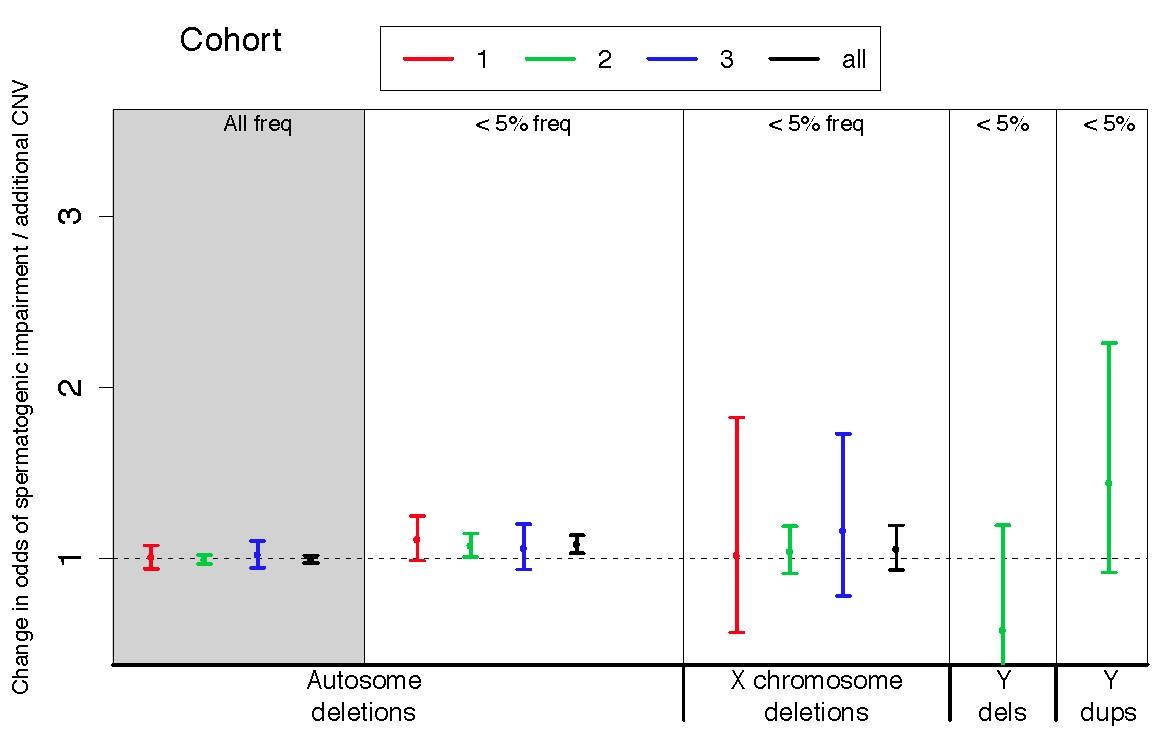
Figure S5: CNV burden statistics using a Spanish control cohort
CNV burden statistics using a Spanish control cohort. In the primary CNV analyses described in the main text, we use a Caucasian population from the United Kingdom as a control group for the Porto azoospermia cohort. Here, we address the effect of using a control group that is more closely matched on genetic ancestry. We performed the same burden analyses depicted in Figure 1 of the main text, this time using a much smaller control cohort of 368 Caucasian men ascertained in Spain. We used logistic regression to estimate the influence of copy number variants (CNVs) on the odds of being diagnosed with impaired spermatogenesis in three case-control cohorts. Eigenvectors from a principal components analysis were used as covariates as before. The odds ratio estimated from fitting a logistic regression model of total CNV count to disease status is plotted separately for each cohort, as well as the combined set of all cohorts (black points). Cohort 1 = Utah (Illumina 370K), 2 = Porto and Weill Cornell (Affymetrix 6.0), 3 = WUSTL (Illumina OmniExpress). Sample sizes used in CNV analysis are n = 83 cases and n = 62 controls for cohort 1, n = 179 cases and 368 controls for cohort 2, and n = 61 cases and 100 controls for cohort 3. Conclusion: While the direction of burden effect for rare autosomal deletions, X-linked deletions, and Y duplications was the same as seen with the analysis using UK controls, only the rare autosomal deletion burden model shows statistical evidence for an odds ratio greater than 1.
Licensed under: https://creativecommons.org/licenses/by/4.0/
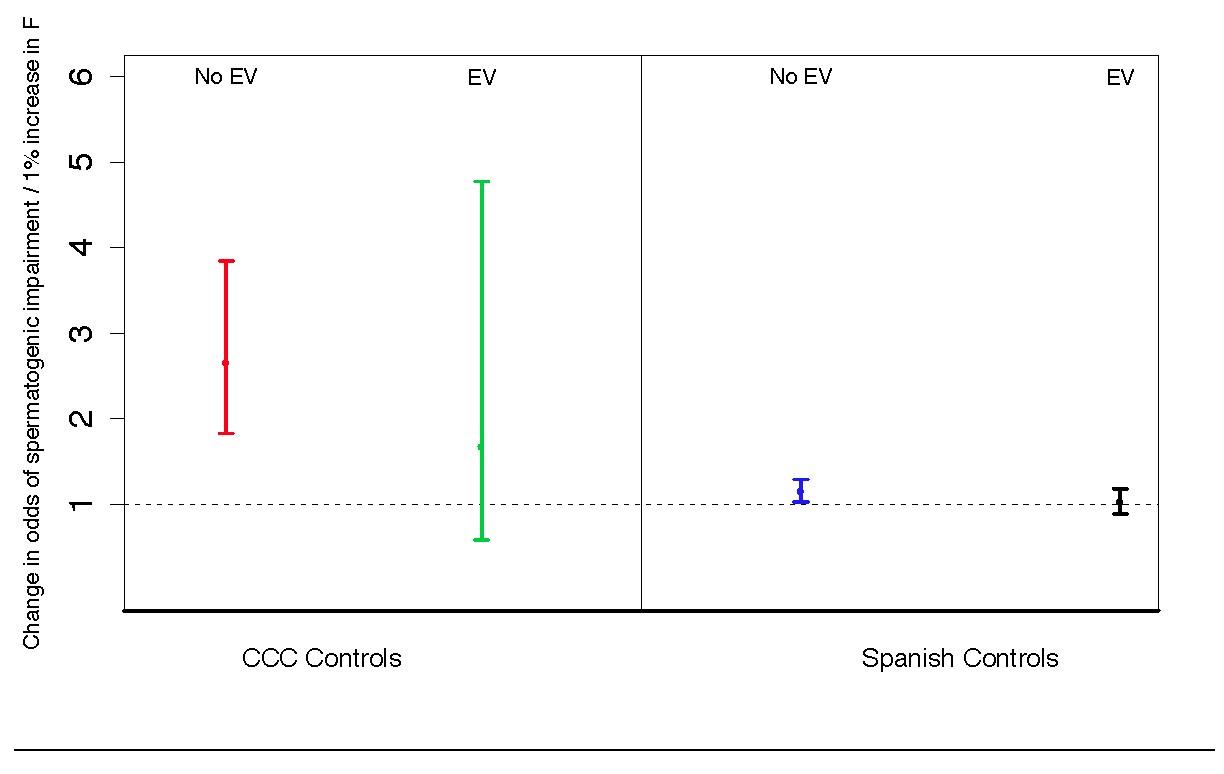
Figure S13: Principal components analysis to assess population structure in case and control cohort
Controlling for population structure while testing for association between inbreeding and infertility. As described in Figure S2 and in Text S1, we used principal components analysis to assess population structure in our case and control cohorts. We use the BEAGLE software package to define homozygous-by-descent regions (HBDRs) of each sample in our study, and used these HBDRs to estimate corresponding inbreeding coefficients. We tested for association between inbreeding coefficient and the probability of azoospermia using a linear model. On the left, we show that there is strong association when analyzing data from the Porto cases and NBS Wellcome Trust case-control consortium controls (CCC controls) in a simple model that does not account for population structure (“No EV”). When including the first 10 eigenvectors from a PCA analysis (“EV”), the point estimate of association remains somewhat inflated but is no longer significant. On the right, we show the same analysis performed on the Porto cohort with a more closely matched control population from Spain, with clearly smaller confidence intervals and smaller point estimates of effect size. In order to be as conservative as possible, we report results of inbreeding analysis using the Spanish control cohort in the main text.
Licensed under: https://creativecommons.org/licenses/by/4.0/
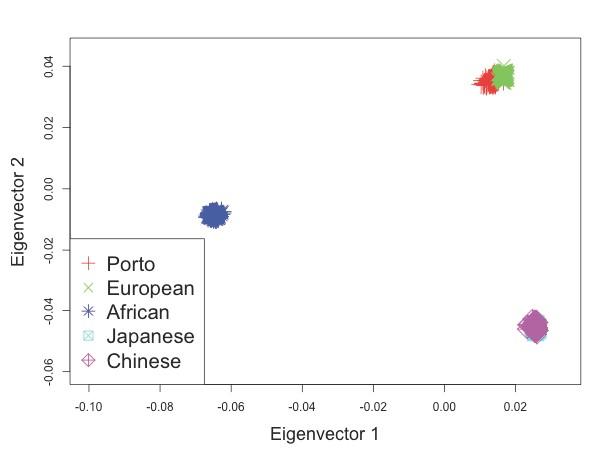
Figure S2 D: Principal components analysis (PCA) of population structure in all Porto cohort post-QC.
Samples were analyzed together with HapMap samples using the EIGENSOFT package [69]. Eigenvector loadings for cases and controls are plotted as red crosses, while HapMap samples are plotted as other colored symbols described in each legend.
Licensed under: https://creativecommons.org/licenses/by/4.0/
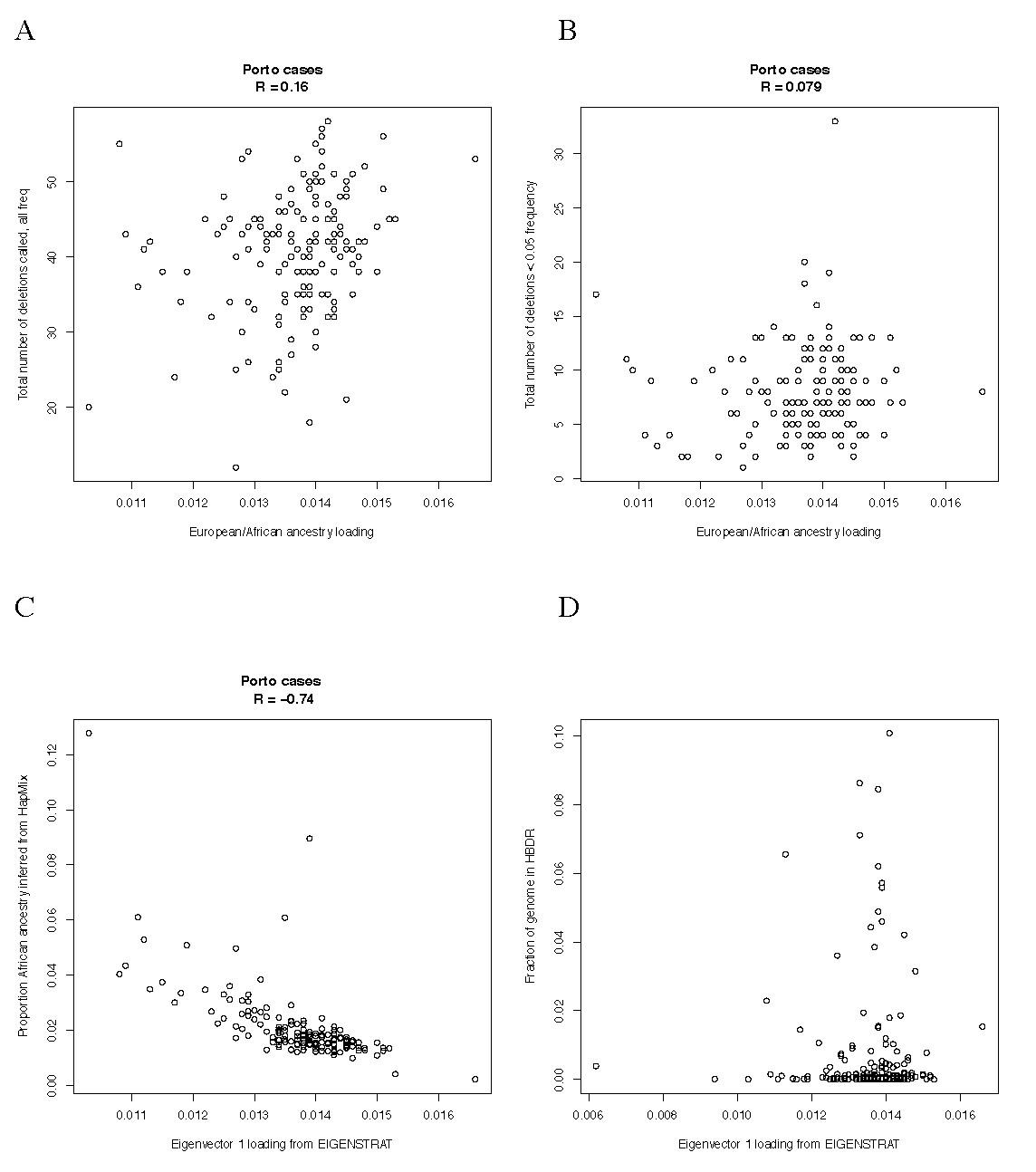
Figure S3: Analysis of population structure in the Porto cohort after sample QC
Based on the results of PCA analysis in Figure S2D, which indicate that the Portuguese population may have subtle differences in allele frequencies from northern European populations, we further investigated the possibility of population structure as a confounder. No significant correlation was observed between the estimated amount of African ancestry in each Porto case and (A) the total number of deletion calls or (B) the total number of rare deletions (here defined as <5% frequency). In both cases smaller (more negative) eigenvector loadings (x-axis) indicate a larger degree of African admixture. We segmented the genome of each sample into regions with 0, 1 or 2 chromosomes of African ancestry using the program HapMix [70]. (C) The percent ancestry inferred by HapMix in this way correlated well with PCA based ancestry estimates. (D) The extent of African ancestry in each case, as estimated by EIGENSTRAT, was uncorrelated (R = 0.002) with the fraction of the genome contained in a homozygous-by-descent region (a rough measure of the inbreeding coefficient), indicating that variation in distant African ancestry was not a major confounder of the HBD analyses.
Licensed under: https://creativecommons.org/licenses/by/4.0/
Data set 3: Replication of the initial findings (Cohort 3- WUSTL)
Genome: Genome-wide Association Study
Species
| Species |
|---|
| Human |
Conditions
| Human phenotype ontology | Participants | Comment |
|---|---|---|
| HP:0003251: Male infertility | 61 | Caucasian men with severe spermatogenic impairment |
| HP:control | 100 | ethnicity-matched, unphenotyped |
Tissue Types
| BRENDA tissue ontology | Maturity | Description | Species | Replicates |
|---|---|---|---|---|
| BTO_0000089: blood | Adult | Human |
Cell Types
| Cell ontology | Maturity | Description | Species | Replicates | Cells per replicate |
|---|---|---|---|---|---|
| CL_0000542: Lymphocytes | Adult | Peripheral blood lymphocytes | Human |
Images
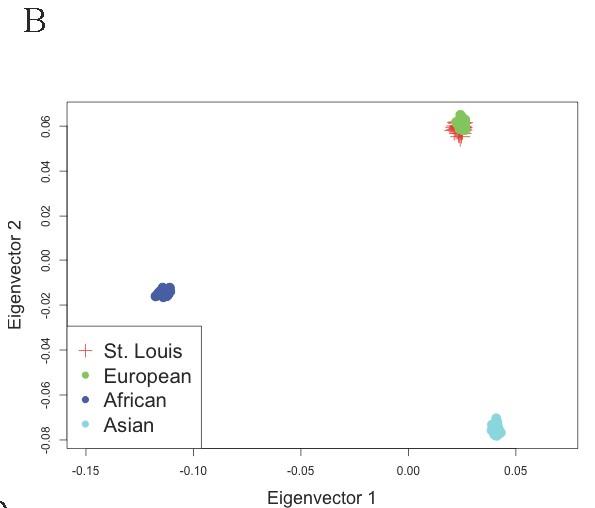
Figure S2 B: Principal components analysis (PCA) of population structure in WUSTL cohort (batch1) post-QC
Samples were analyzed together with HapMap samples using the EIGENSOFT package. Eigenvector loadings for cases and controls are plotted as red crosses, while HapMap samples are plotted as other colored symbols described in each legend.
Licensed under: https://creativecommons.org/licenses/by/4.0/
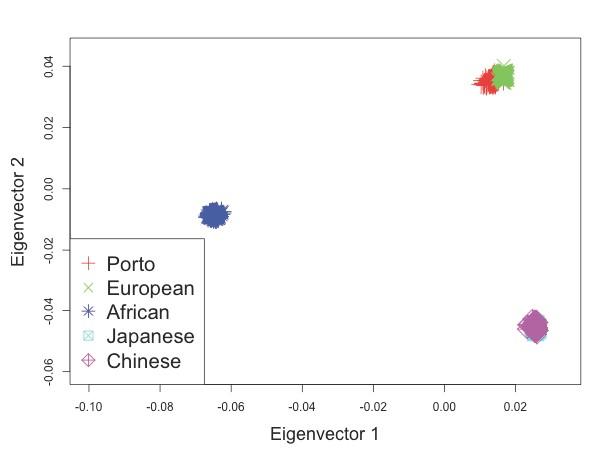
Figure S2 C: : Principal components analysis (PCA) of population structure in WUSTL cohort (batch 2) post-QC
Samples were analyzed together with HapMap samples using the EIGENSOFT package. Eigenvector loadings for cases and controls are plotted as red crosses, while HapMap samples are plotted as other colored symbols described in each legend.
Licensed under: https://creativecommons.org/licenses/by/4.0/
Data set 4: Comparison between Cohort 1, 2 and 3
Genome: Genome-wide Association Study
Species
| Species |
|---|
| Human |
Conditions
| Human phenotype ontology | Participants | Comment |
|---|---|---|
| HP:0011961: Non-obstructive azoospermia | 35.0 | men from Utah (Cohort 1) |
| HP:0000027: Azoospermia | 179.0 | men from Porto, Weill Cornell (Cohort 2) |
| HP:0000798: Oligozoospermia | 48.0 | men from Utah (Cohort 1) |
| HP:0003251: Male infertility | 61.0 | Washington University, WUSTL (Cohort 3) |
| HP:control | 62 men from Utah (Cohort 1); 368 men from Spain (Cohort 2); 100 men WUSTL (Cohort 3) |
Tissue Types
| BRENDA tissue ontology | Maturity | Description | Species | Replicates |
|---|---|---|---|---|
| BTO_0000089: blood | Adult | Human |
Cell Types
| Cell ontology | Maturity | Description | Species | Replicates | Cells per replicate |
|---|---|---|---|---|---|
| CL_0000542: lymphocyte | Adult | Peripheral blood lymphocytes | Human |
Images
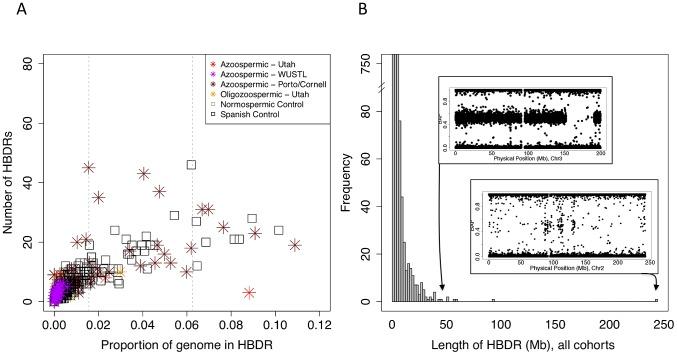
Figure 4: Patterns of homozygosity in men with low sperm count
(A) Distribution of the number of HBD regions (HBDRs), and the proportion of genome contained in these putative HBD regions, plotted for each sample in this study. Replication case and control cohorts are indicated in the legend.<br/>(B) Length distribution of HBDRs detected in all samples combined. Inset, two panels showing probe level intensity data corresponding to the two largest HBDRs detected. BAF: b-allele frequency, calculated as B/(A+B) where A and B are the approximate copy numbers for the A and B allele, respectively. The largest HBDR detected corresponds to a case of uniparental disomy of chromosome 2 (UPD2) detected in an azoospermic man from the Utah cohort.
Licensed under: https://creativecommons.org/licenses/by/4.0/
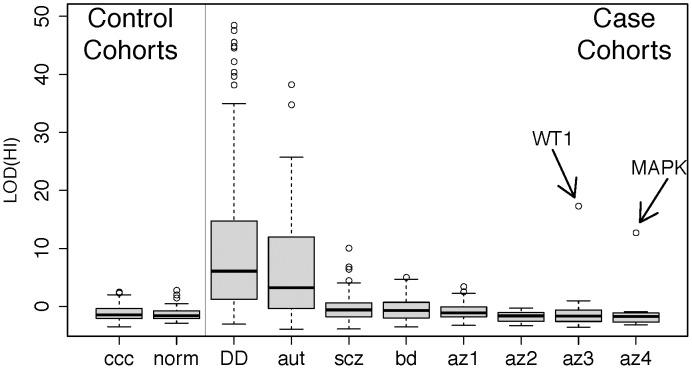
Figure 3: Disruption of predicted haploinsufficient genes is infrequent in spermatogenic failure
We obtained lists of rare deletions, left panel, from the Utah and WTCCC control cohorts and, right panel, from cohorts of developmental delay (DECIPHER) [66], autism [67], schizophrenia [68], bipolar disorder [66], [68], and spermatogenic impairment (this study). We used a published method for assessing the likelihood that each deletion disrupts a haploinsufficient gene [47], summarized as a LOD score, and ordered each cohort by the median LOD(HI) within cases and controls separately. While the CNVS from DECIPHER (p<1×10−15), autism (p<1×10−15), schizophrenia (p<1×10−4) and bipolar disorder (p<0.002) show significant enrichment of high LOD (HI) scores compared to controls, the infertility cohorts have score distributions indistinguishable from controls. Two outlier deletions from the infertility cohort are annotated; one is a deletion of WT1, a key gene in gonadal differentiation, and the other is a 1 Mb deletion involving several genes including MAPK1 and the cancer-testis antigen PRAME. Further review of clinical data from the WT1 carrier showed signs of cryptorchidism. Abbreviation of azoospermia cohorts: az1, Utah cohort, az2, WUSTL, az3 Porto, az4, Weill-Cornell. Note that for additional detail we have split the cohort referred to as “Porto” in the main text into two subgroups, az3 and az4, defined by the clinical group that ascertained the cases
Licensed under: https://creativecommons.org/licenses/by/4.0/
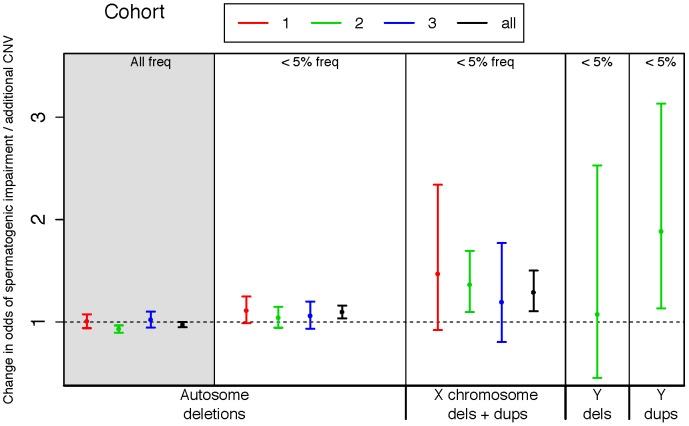
Figure 1: Rare variant burden in cases of spermatogenic impairment
We used logistic regression to estimate the influence of copy number variants (CNVs) on the odds of being diagnosed with impaired spermatogenesis in three case-control cohorts. The estimated odds of spermatogenic impairment is equal to, or slightly lower than, one when considering autosomal deletions of all frequencies (leftmost panel, shaded grey). However, when considering only autosomal deletions with call frequencies less than 5%, we observed a progressively increasing risk conferred by events on the autosomes, the X and the Y chromosomes. A very small number of Y-linked calls were made in cohorts 1 and 3 due to array design, thus we have only plotted Y-linked rates for cohort 2. Samples with Y-linked AZF deletions were excluded from the study. The odds ratio estimated from fitting a logistic regression model of total CNV count to disease status is plotted separately for each cohort, as well as the combined set of all cohorts (black points). Cohort 1 = Utah (Illumina 370K), 2 = Porto and Weill Cornell (Affymetrix 6.0), 3 = WUSTL (Illumina OmniExpress), All = meta-analysis of all three cohorts. Sample sizes used in CNV analysis are n = 83 cases and 62 controls for cohort 1, n = 183 cases and 974 controls for cohort 2, and n = 61 cases and 100 controls for cohort 3.
Licensed under: https://creativecommons.org/licenses/by/4.0/
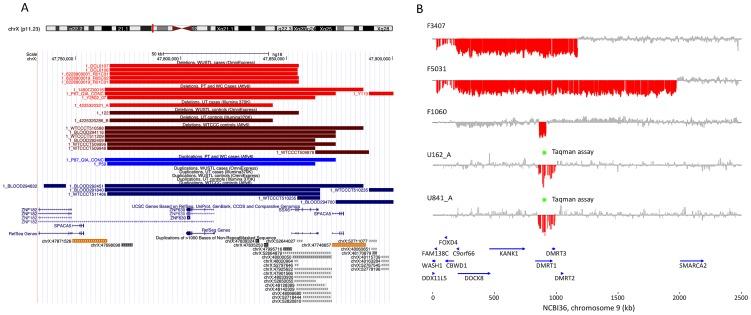
Figure 2: Discovery of recurrent deletions in azoospermia
(A) A recurrent microdeletion on Xp11.23 (47765109–47871527 bp, hg18) is a strong candidate risk factor for spermatogenic failure. The location of deletions (red shades) and duplications (blue shades) in cases and controls are plotted separately for each cohort. CNVs at this locus appear to arise due to non-allelic homologous recombination between two nearly identical (>99.5% homology) 16 kb segmental duplications that contain the sperm acrosome gene SPACA5. Also within the CNV region are the genes ZNF630 and the cancer-testis antigen SSX6. We identified 9 deletions of this locus spread across all patient cohorts (3 in PT, 1 in UT, 5 in WUSTL) compared to 8 in the pooled 1124 controls (2.8% frequency versus 0.7%, odds ratio = 3.96, p = 0.005, Fisher exact test). After analysis of an additional 403 cases and 2121 controls, the association is still significant (combined data: 1.6% frequency in cases, 0.55% in controls, OR 3.0, 95% CI = [1.31–6.62], p = 0.007). (B) We identified two patients with deletion of DMRT1, a gene on 9p24.3 that is orthologous to the putative sex determination locus of the avian ZW chromosome system [36]. Both men were diagnosed as azoospermic. We validated these deletion calls with a qPCR assay (green star, Figure S9). We screened Affymetrix 6.0 data from an independent Han Chinese case-control study of NOA and identified an additional 3 deletions of DMRT1 coding sequence in 979 cases and none in 1734 controls. Finally, we observed no coding deletions of DMRT1 in the two largest control SNP array datasets in the Database of Genomic Variants, consisting of 4519 samples [42], [43]. The combined results indicate that deletion of DMRT1 is a highly penetrant genetic cause of human spermatogenic failure (frequency of 0.38% in 1306 cases and 0% in 7754 controls, combined p = 6.2×10−5). Patient IDs are indicated next to each plot (U162_A, U841_A = Utah cohort patients; F3407, F5031, F1060 = Nanjing cohort patients).
Licensed under: https://creativecommons.org/licenses/by/4.0/

Figure S9: CNV calls made at the DYZ19 tandem repeat locus
CNV calls made on the Utah, Porto, and WUSTL case cohorts, and the Utah, NBS (WTCCC), WUSTL and Spanish control cohorts.
Licensed under: https://creativecommons.org/licenses/by/4.0/
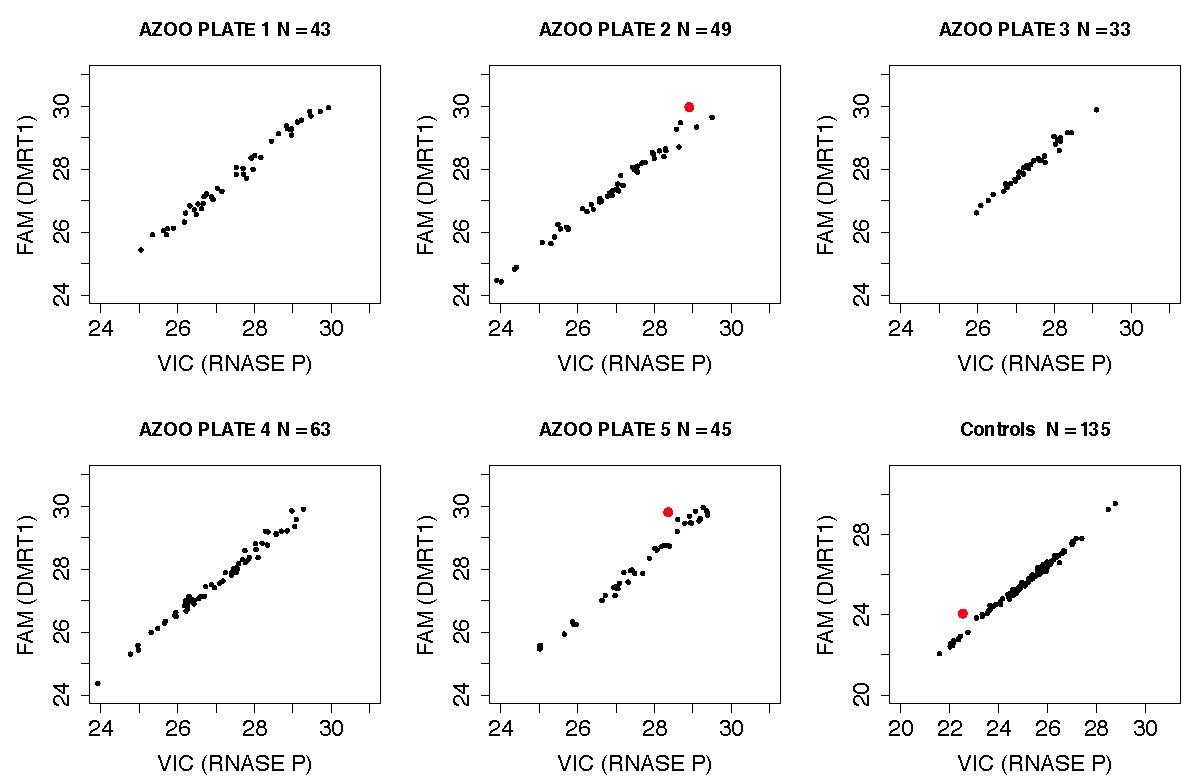
Figure S12: Detection of additional intronic DMRT1 deletions using the TaqMan validation assay
As described in the supplemental methods, we screened 5 plates of DNA from Weill Cornell cases and 2 plates of Caucasian male controls using the DMRT1 TaqMan validation assay. Each sample was assayed in quadruplicate (although some samples were assayed in duplicate or triplicate if insufficient DNA was available). We applied extremely stringent calling criteria to these data, excluding samples with (1) low DNA content as defined by picogreen assay (2) high standard deviation (>0.3) of delta CT measurements across replicates (3) low copy number confidence scores generated by CopyCaller software. Each point in the panels above represents the average delta CT for the control locus (VIC) and DMRT1 (FAM) for a single sample. Red dots indicate deletion carrier calls. We detected 2 deletions in 233 case samples, a frequency of 0.86%, and 1 deletion in 135 controls (0.74%). As this assay is targeting intronic sequence, and we have not cloned the breakpoints the functional consequences of each deletion is unclear.
Licensed under: https://creativecommons.org/licenses/by/4.0/
Data set 5: HBD analysis- Identification of a candidate azoospermia mutation in the case of UPD2.
Exome: Whole Exome Sequencing
Species
| Species |
|---|
| Human |
Images
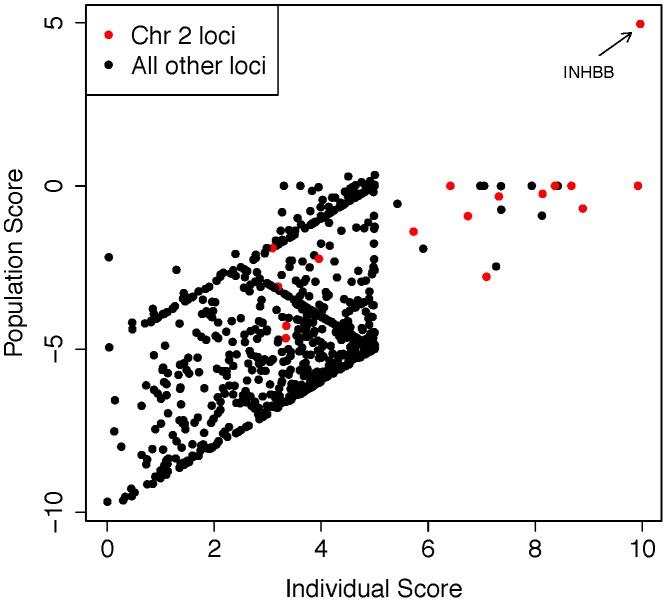
Figure 5: Analysis of exome sequencing data identifies a candidate azoospermia mutation in the case of UPD2.
We performed whole-exome sequencing on the case of UPD2 in an attempt to identify a potential genetic cause for this mans azoospermia. We constructed a scoring method to rank order the exome variants in two dimensions: (i) within the set of variants seen in this single exome, the “Individual Score” and (ii) across a large set of exome sequences, the “Population Score”. For each exome variant, the Individual Score, Pind,, was constructed by summing normalized predictions of functional impact from 5 commonly used annotation algorithms: PhyloP, PolyPhen2, SIFT, GERP, and LRT. This score was then multiplied by the ploidy of the mutant allele (e.g. 1× for a heterozygous genotype and 2× for a homozygous genotype) creating a final Individual Score ranging from 0–10. We also calculated the Individual Score for all variation in the 1000 genomes Phase I sequencing data. To construct the “Population Score” for each variant in the UPD individual, Ppop, we identified the maximum Individual Score variant in the corresponding gene, Pmax, within the 1000 genomes data, and defined Ppop = Pind−Pmax. The purpose of the Population Score is to scale the importance of each Individual Score by the extent of pathogenic variation that exists in the population at each gene. Only sites with minor allele frequencies less than 10% in both the 1000 genomes data and the Exome Variant Server (http://evs.gs.washington.edu/EVS/) were considered in the analysis. When examining the joint distribution of Ppop and Pind for the UPD2 individual, we saw an enrichment of large scores for variants on chromosome 2, as expected. The most extreme variant on both scales was a homozygous nonsense mutation in the gene INHBB, the implications of which we discuss in Figure 6.
Licensed under: https://creativecommons.org/licenses/by/4.0/
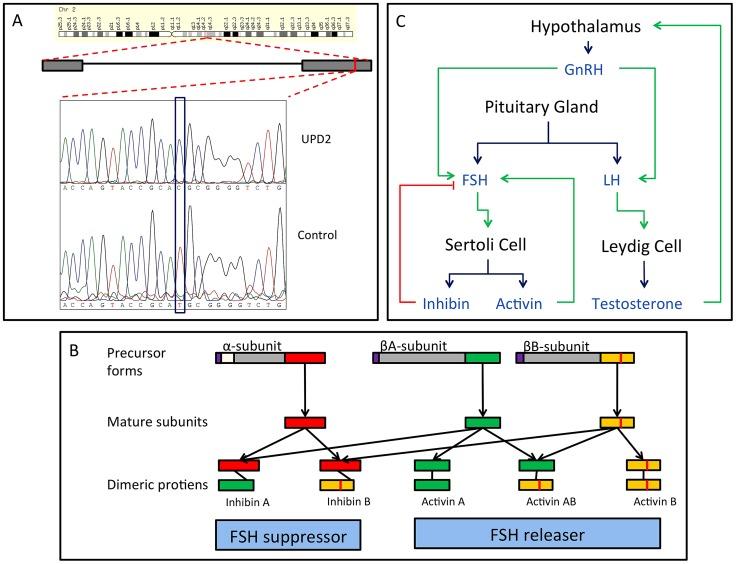
Figure 6: Homozygous missense mutation of INHBB identified in the case of UPD2.
A: We validated this candidate by Sanger sequencing in the UPD2 case and control individuals. Mutant and reference nucleotides are highlighted within the blue box, confirming the homozygous T to C nucleotide change observed at chr2:12,1107,305 bp (hg19) of the UPD2 individual. Grey boxes represent the exons of the gene and the red line indicates the location of the observed mutation within the gene.<br/>B: INHBB encodes for the protein, Inhibin βB, which along with inhibin α and inhibin βA, combine combinatorially to form the inhibins and activins. Each protein expressed by INHA, INHBA, INHBB consists of an N-terminal signal peptide (purple), a propeptide (grey), and a subunit chain (green, red or yellow). The mutation identified here results in a M370T change of the inhibin βB subunit chain (location indicated by a vertical red line throughout the diagram). The various inhibin subunits dimerize via disulfide bonds (locations indicated by black lines between subunits). As the βB subunit participates in multiple complexes with antagonistic functions, the functional consequences of loss-of-function or gain-of-function mutations in this protein may be difficult to predict.<br/>C: The role of inhibins and activins in the hypothalamic-pituitary testicular axis. These complexes have diverse functions in the body, but are most well known for their ability to stimulate and inhibit follicle stimulating hormone (FSH) production, a process critical for spermatogenesis. Blue arrows connect hormones to the cell or gland by which they are secreted. Green arrows indicate stimulatory interactions, and red lines indicate inhibitory interactions.
Licensed under: https://creativecommons.org/licenses/by/4.0/